Parkinson’s Disease (PD)
- Parkinson’s disease (PD) is the most common movement disorder4
- 2nd most common neurodegenerative disease, after Alzheimer’s disease4
Epidemiology
2nd most prevalent progressive neurodegenerative disorder after Alzheimer’s Disease5
Etiology
Most cases of PD are likely the result of a combination of environmental and genetic factors4
Primary PD (idiopathic)
Secondary PD
Parkinson-Plus
Genetic
- Heritability of PD6:
- Using genome wide complex trait analysis, studies have found even higher percent estimates of 22-40% of PD cases having a genetic component → capturing both highly penetrant monogenic forms/cumulative effect of common variants8.
- 22-40% of PD cases have genetic factors that contributed to developing their PD
- Just because one has some genetic predisposition to PD does NOT mean they will get PD
- There are gene-environment interactions: (with outside influences like pesticides, tobacco use, chemicals etc) which can exacerbate genetic susceptibility to developing the disease8.
A Family with an autosomal dominant Parkinson’s disease allowed the discovery of a mutation in the SNCA gene → leading to identification of alpha synuclein protein as a hallmark component of lewy bodies/neurites in PD8.
- Pathogenesis of DJ-1/PARK7-Mediated Parkinson’s Disease9
Neuroinflammation
Sub types
Secondary Parkinsonism
- Postencephalitic Parkinsonism
- Toxic Parkinsonism
- Drug-Induced Parkinsonism
Postencephalitic Parkinsonism11
- Viral brain infection
- Not seen today
Toxic Parkinsonism(O’Sullivan, Schmitz, and Fulk 2019)
Exposure to environmental toxins Pesticides Industrial chemicals (manganese, CO, cyanide, methanol) Synthetic heroin + MPTP
Drug-Induced Parkinsonism
Drug-Induced Parkinsonism11 - Drugs that result in extra-pyramidal dysfunction → Pseudo PD symptoms - Neuroleptic drugs: (chlorpromazine) (Thorazine®), haloperidol (Haldol®), thioridazine (Mellaril®), and thiothixene (Navane®) - Antidepressant drugs: amitriptyline (Triavil®), amoxapine (Asendin®), and trazodone (Desyrel®) - Antihypertensive drugs: Methyldopa (Aldomet®) and reserpine
Metabolic PD
- PD d/t metabolic conditions
- Rare
- Calcium metabolism dysfunction → BG Calcification
- Hypothyroidism
- Hyperparathyroidism
- Wilson’s Disease
Parkinson-Plus
Parkinson-Plus refers to a group of neurodegenerative diseases can affect the substantia nigra and produce parkinsonian symptoms along with other neurological signs11
Pathologies
- Cortical–basal ganglionic degeneration (CBGD)
- Progressive supranuclear palsy (PSP)
- Multiple system atrophy (MSA) syndromes (striatonigral degeneration [SND])
- Shy-Drager syndrome
- Sporadic olivopontocerebellar atrophy [OPCA]
- Motor neuron disease parkinsonism
Parkinson-like symptoms
- Multi-infarct vascular disease
- Dementia syndromes (Alzheimer’s disease, diffuse Lewy body disease [DLBD], and frontotemporal dementia [FTD])
- Normal pressure hydrocephalus (NPH)
- Creutzfeldt-Jakob disease (CJD)
- Wilson’s disease (WD)
- Juvenile Huntington’s disease
Pathophysiology
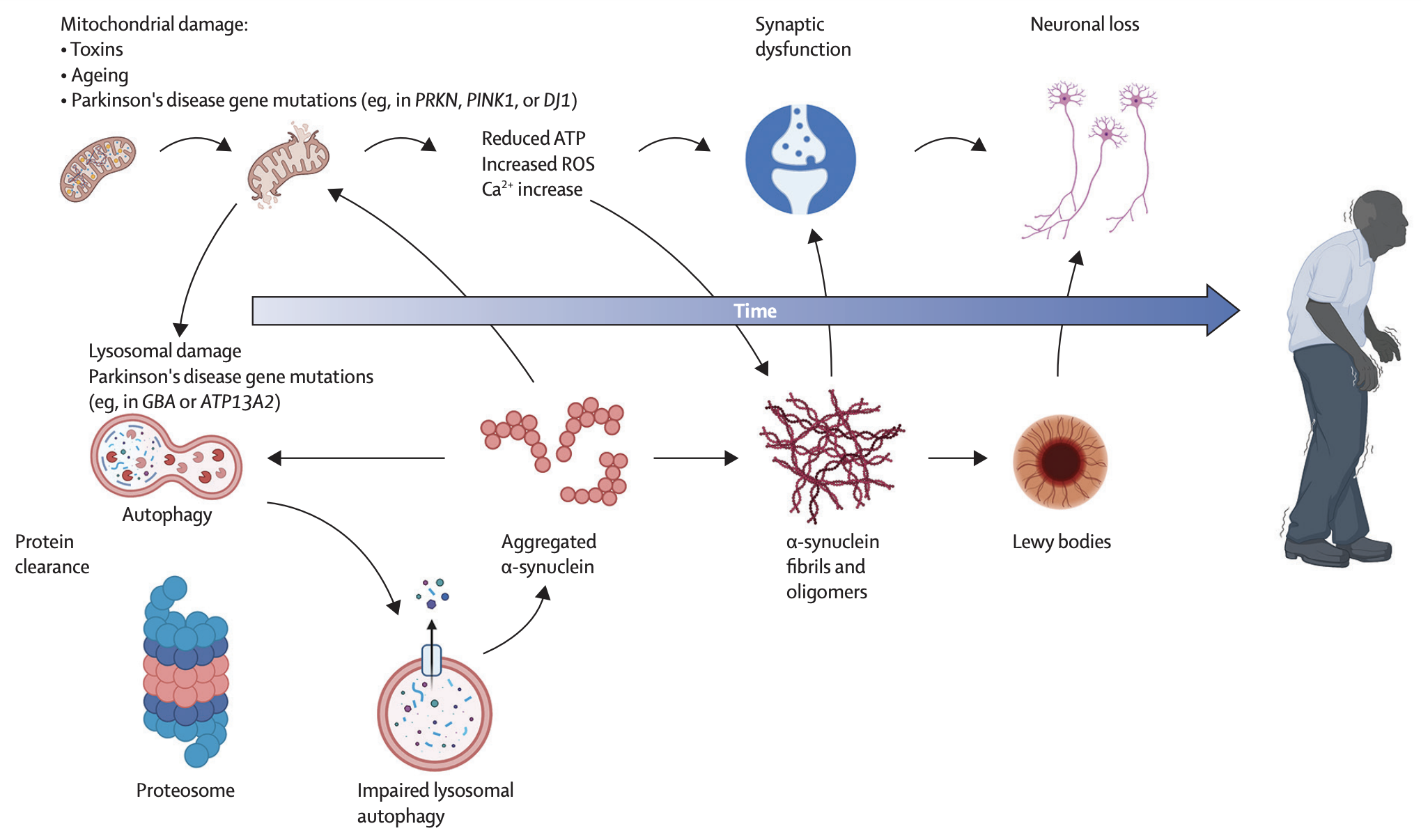
Note
“Physical manifestation of lewy bodies in the brain, whereas Dementia is the cognitive manifestation of lewy bodies in the brain” -Adria Thompson
Primary disturbances in the dopamine systems of basal ganglia (BG)11
The pathophysiology of PD is characterized by 2 phases11
- Degeneration of dopaminergic neurons in the SNpc11
- Addition of Lewy Bodies11
| Proposed pathogenesis | Genetic evidence | Biomarkers | Therapeutic implications | |
|---|---|---|---|---|
| ↑ SNCA expression | ↑ in α-syn protein results in increased aggregation in cell death and dysfunction | ↑ SNCA gene dose results in PD | α-syn and phospho-syn blood and CSF measures | ↓ in SNCA transcription or translation |
| ↑ α-syn aggregation | Formation of oligomers and fibrils is toxic to cells | Coding mutations in SNCA gene lead to α-syn aggregation | rt-QUIC assays of CSF, skin and olfactory mucosal biopsies | Anti-aggregation therapies |
| Mitochondrial dysfunction |
Reduced complex 1 activity abnormal calcium homeostasis increased reactive oxygen species reduced mitochondrial ATP production |
Multiple PD gene mutations lead to changes in mitochondrial function e.g. PRKN, PINK1 and LRRK2 |
Magnetic resonance spectroscopy analysis of Pi/ATP ratios; Measurement of ATP and mitochondrial function in skin biopsies |
Enhancing mitochondrial biogenesis and function |
| Altered endosomal-lysosomal trafficking | Activation of LRRK2 and VPS35 lead to phosphorylation of RAB proteins leading to ↓ lysosomal function; and altered response to membrane damage | Rare pathogenic variants in LRRK2 (e.g. G2019S) and VPS35 lead to ↑ RAB phosphorylation | Serum Rab protein phosphorylation | Reducing LRRK2 protein levels and/or kinase activity via ASO therapy or kinase inhibitors |
| Lysosomal dysfunction | Impaired α-synuclein degradation leading to increased cellular α-synuclein |
GBA1 mutations are associated with PD; rare variants in other genes may be relevant |
GCase protein and enzyme activity measurements; GSLs in blood and CSF |
Modulators of β-glucocerebrosidase activity |
| Immune activation & neuroinflammation | Multiple factors (α-syn aggregates, mitochondrial antigens, and gut endotoxins) trigger immune responses, driving neuroinflammation and neuronal toxicity |
HLA variants are associated with PD; LRRK2, PRKN and PINK1 are involved in inflammatory pathways |
C-Reactive protein interleukins PET imaging of activated microglia |
Immunomodulatory or anti-inflammatory therapies |
| Cell to cell spread | Toxic α-syn can spread to neighboring and distant cells via extracellular vesicles (potentially) | N/a | α-syn aggregations via rt-QUIC assays of CSF, skin, and olfactory mucosal biopsies |
Monoclonal antibodies or other therapies: ↓ release of toxic proteins Inhibition of extracellular transit ↓ reuptake by recipient cells |
| Source: Morris et al. 20248 | ||||
Degeneration of Dopaminergic neurons
- Loss of the melanin-containing neurons produces characteristic results in Depigmentation in the substantia nigra with a characteristic pallor11.
- Numerous other brain regions of people with PD show structural and functional changes resulting in impaired modulation of other neurotransmitters (acetylcholine, serotonin, noradrenaline, glutamate, and GABA)11.
α-Synuclein & Lewy bodies
In normal systems, α-synuclein is an amino acid protein found in brain abundantly in the synaptic terminals of neurons to help facilitate vesicle transport/neurotransmitter release8.
When dysfunction occurs, α-synuclein can aggregate into clumps known as lewy bodies8.
- Detrimental to neurons- they are space occupying lesions that could alter cellular function
- Braak + colleagues used alpha syn staining to detect Lewy bodies post mortem
- Found PD can start in gut or olfactory system then spread to cortical/subcortical brain regions
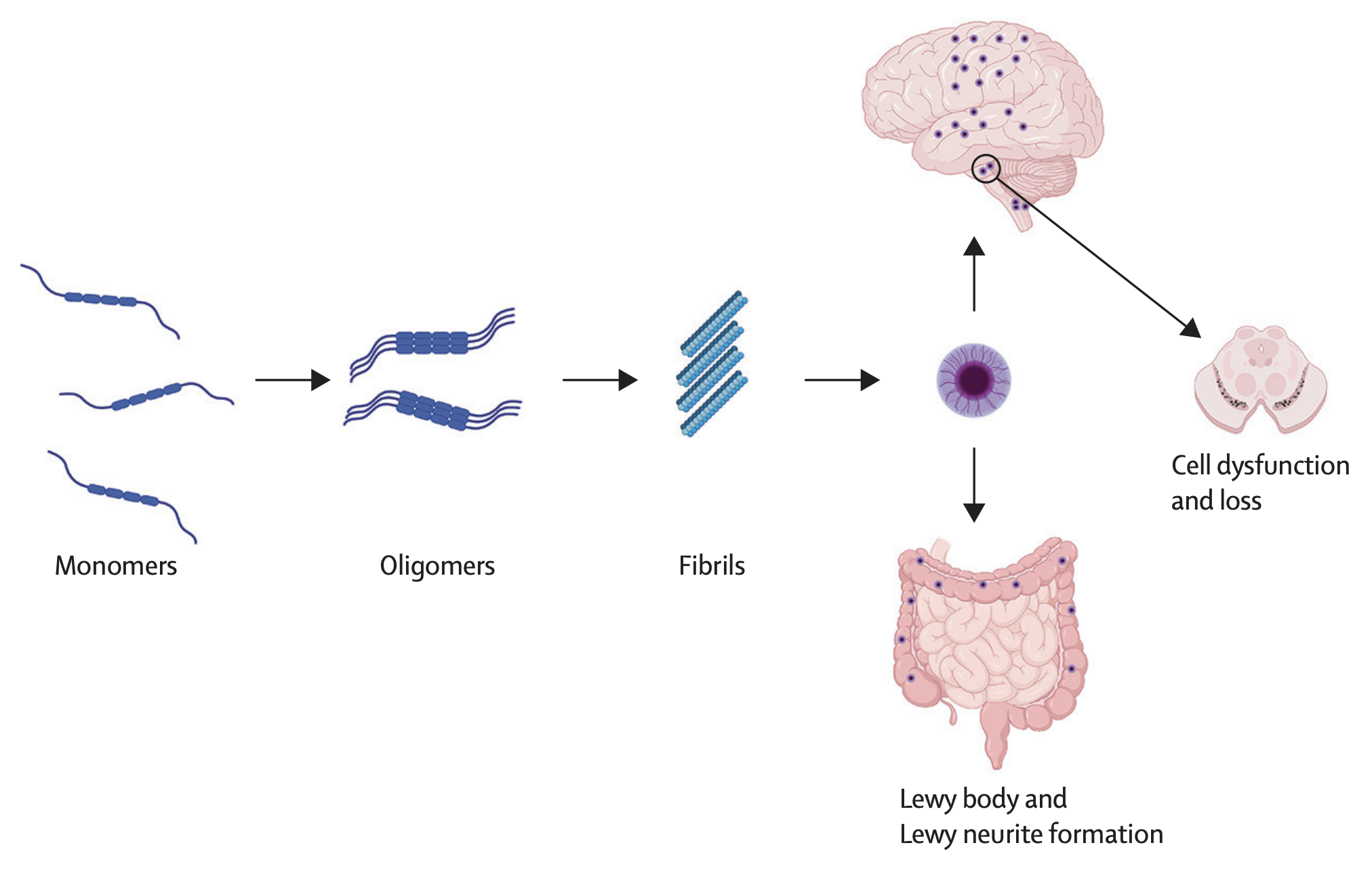
Note
While presence of Lewy bodies is a hallmark of Parkinson’s disease, they are not universally found in all patients with the condition. This highlights the heterogeneity of PD and the complexity of its underlying pathophysiology8.
Some PD cases start in brainstem or limbic system or other areas Hypothesis of spreading alpha syn has been validated in multiple studies: Injection of alpha syn fibrils into brain leading to aggregation found in post mortem PD (performed in mice, primates) Found A-Syn can spread from periphery to CNS or from brain to other organs
Changes are seen in the pedunculopontine nucleus (PPN) and nucleus basalis of Meynert (nbM) that release acetylcholine (ACh) Degeneration & Alpha Synuclein deposition (early in PD) at Locus coeruleus that releases noradrenaline
Gut-Brain Axis
The relationship between the gut brain axis and pathogenesis of Parkinson’s Disease was suggested in Braak’s hypothesis4
- This hypothesis builds upon that idea that in PD, α-synclein aggregates in other areas in the body, not just the substantia nigra (the hallmark site)
- Braak’s Hypothesis places emphasis on the gastrointestinal tract4.
Animal studies
Researchers have found a bidirectional spread of α-syn from the duodenum to the brainstem and the stomach after injecting α-syn in rats4
The fact that 20% of patients experience constipation prior to motor symptoms could be evidence that the gut-brain axis plays a role in the mechanism of Parkinson’s Disease12.
Mitochondrial pathomechanisms
Mitochondria, the powerhouse of the cell, play a key role in cellular energy production/cell signalling8. Mitochondrial dysfunction is proposed to be an early event in pathogenesis of PD8. Alterations of mitochondrial structure/dynamics linked to increased production of reactive oxidative species (ROS), abnormal intracellular calcium levels, and reduced ATP production8.
The results of the following studies supported this pathological mechanism:
- Inhibition of mitochondrial complex 1 induces PD like symptoms in animal models8
- Mitochondrial dysfunction can result in production of ROS → leading to oxidative stress/neuronal damage8
- Mitochondrial DNA mutations have been implicated in risk of developing PD8
- Alpha Syn aggregation can impair mitochondrial function8
- Decreased mitochondrial complex 1 activity has been reported in PD8.
Lysosomes
- PD ⇒ could involve a decline in clearance capacity of autophagy-lysosomal systems or ubiquitin-proteasome systems8.
- Lysosomes are involved in autophagy- they clean out abnormal/accumulated proteins8.
- α-syn degradation is lysosomal dependent8.
- If lysosomes are impaired, this would affect A-syn turnover, causing an aggregation of &alpha-syn into lewy bodies8.
- Mutations in lysosomal pathway genes have been shown to result in increased a syn accumulation which could lead/progress PD8.
Caution
This lysosome pathophysiology is not fully straightforward and the mechanisms are not fully understood8.
Auto-immune & Inflammatory Mechanisms
In post-mortem studies, microglial activation/ elevated inflammatory cytokines found in post-mortem brains of those with PD8.
- Found in blood low grade elevation of inflammatory cytokines → linked to rapid disease progression8.
- Immune activation has different roles that could give benefit → particularly in early stages of neurodegeneration with promoting clearance of abnormal protein aggregates8.
- Later on, dysfunction of immune mediated clearance mechanisms could cause more a-syn aggregate accumulation although if this is protective to balance shifts is yet to be explored8.
- Is immune activation is a primary determinant of disease progression or a secondary phenomenon? This is a debate8.
- Lots of evidence to support that it is actually a Primary determinant8.
- Monogenic causes of PD are linked to the immune system (LRRK2 gene)8.
- Use of immunosuppressants/corticosteroids are associated with reduced PD risk8.
- Changes in gut microbiome can cause the pro inflammatory species found in PD8.
- Gut inflammation can cause 3 potential mechanisms or a combo:8.
- → gut leakage of inflammatory mediators to blood/BBB (thru blood brain barrier),8.
- →A syn aggregation promoted in enteric neurons travels via vagus nerve to brain8.
- → A syn T cell response in gut w trafficking of these T cells to sites of alpha syn pathology in brain8.
- Is immune activation is a primary determinant of disease progression or a secondary phenomenon? This is a debate8.
Insulin Insensitivity
Insulin resistance + impaired glucose metabolism are associated with the development and progression of PD13. Insulin resistances promotes PD through increased α-syn expression, mitochondrial dysfunction, and elevated ROS production13.
In 2023, Ruiz-Pozo et al.14, performd a systematic review investigating the relationship between insulin resistance and Parkinson’s Disease. Insulin resistance was associated with α-syn aggregation, dopaminergic neuronal loss, autophagy, and neuroinflammation14.
Based on this information, this can be helpful in both risk assessment and treatment.
Blood tests determining insulin sensitivity/metabolic health can be useful in determining risk of developing PD14.
For patients who already have PD, metabolic health and specifically insulin sensitivity should be considered when developing a plan of care. Enhancing insulin signaling pathways can have neuroprotective effects/improve motor and cognitive fxns in PD patients15,16.
Interventions to enhance insulin sensitivity include:
- Balanced diet
- Exercise
- Antidiabetic agents
- Insulin therapies
Exercise - increases glucose uptake in muscles thu insulin independence paths/improves mitochondrial fxn
Diet -
Antidiabetic agents like exenatide, which improve insulin sensitivity, have shown promise in clinical trials for PD, leading to significant improvements in motor and non-motor symptomsBasal Ganglia Loop Dysfunction
Direct Loop
- Consists of signals transmitted from the cortex to putamen to globus pallidus, to ventrolateral (VL) nucleus of the thalamus, and back to cortex (supplementary motor area [SMA]).
- This VL-SMA connection is excitatory and facilitates discharge of cells in the SMA11.
- The BG thus serves to activate the cortex via a positive-feedback loop and assists in the initiation of voluntary movement11
- Inhibition of the thalamus by the BG is thought to underlie the hypokinesia seen in PD11
Indirect Loop
- STn, Gpi, and SNpr → Superior colliculus and midbrain tegmentum11.
- Serves to decrease thalamocortical activation11.
- BG projection to the superior colliculus assists in regulation of saccadic eye movements11.
- The BG projection to the reticular formation assists in the regulation of trunk and limb musculature (via extrapyramidal pathways), sleep and wakefulness, and arousal. Other circuits in the BG are involved with memory and cognitive functions11.
Clinical Presentation
Onset
- It is currently believed that PD begins many years before motor symptoms become clinically relevant, but the non-motor symptoms may appear around onset4
Symptoms
Symptons can be divided into “motor” and “non-motor” symptoms.
Gait Symptoms
- Parkinsonian Gait
- Freezing of gait (FOG)
Diagnosis
Parkinson’s Disease (PD) is defined by the Movement Disorder Society Clinical Criteria: as Bradykinesia with rest tremor, Rigidity, or both17.
Note
These features must be clearly demonstratable, and cannot be due to confounding factors17.
Diagnostic criteria from MDS-PD can place patients into 2 categories:
- Diagnosis of clinically established PD
- Diagnosis of clinically probable PD
Clinically established PD
- Absence of absolute exclusion criteria
- ≥2 supportive criteria
- No red flags
Clinically probable PD
- Absence of absolute exclusion criteria
- Presence of red flags counterbalanced by supportive criteria
Supportive Criteria
If a patient has the “supportive criteria” (need 1 for probable and at least 2 for clinical PD):
- Dramatic beneficial response to dopaminergic therapy (improvement in motor sx)17.
- Levodopa induced dyskinesia17.
- Rest tremor of limb (usually 1 limb)17.
- Loss of olfaction (loss of smell)17.
- Cardiac sympathetic denervation (loss of postganglionic sympathetic nerve fibers, hallmark of PD)17.
- Autonomic dysfunction can lead to OH/Arrhythmias17.
- No exclusion criteria17.
- No red flags17.
Absolute exclusion criteria
- Cerebellar abnormalities: The presence of cerebellar signs such as ataxia, which are not consistent with PD17.
- Downward vertical supranuclear gaze palsy: This includes significant slowing of vertical saccades, which is more indicative of progressive supranuclear palsy (PSP)17.
- Parkinsonian features restricted to the lower limbs for more than three years: This presentation is atypical for PD17.
- Treatment with a dopamine receptor blocker or dopamine-depleting agent: If parkinsonism persists despite discontinuation of the offending drug, it suggests drug-induced parkinsonism rather than PD17.
- Absence of response to high-dose levodopa: If there is no significant improvement in motor symptoms with adequate doses of levodopa, it suggests an alternative diagnosis17.
- Cortical sensory loss, ideomotor apraxia, or progressive aphasia: These features are more consistent with corticobasal degeneration (CBD)17.
- Normal functional neuroimaging of the presynaptic dopaminergic system: This includes normal findings on DaTSCAN or similar imaging, which would be inconsistent with PD17.
- Documentation of an alternative condition known to cause parkinsonism: This includes conditions such as multiple system atrophy (MSA), PSP, or CBD, confirmed by clinical or imaging findings17.
Red flags
“Red flags” as defined by the clinical criteria refer to signs or symptoms that, if observed, If you see these, need to differentiate from atypical PD like MSA, PSP, CBD
- Rapid progression of gait impairment requiring wheelchair use within 5 years of onset17.
- Absence of progression of motor symptoms or signs over 5 years unless stability is related to treatment17.
- Early bulbar dysfunction, such as severe dysphonia or dysarthria within the first 5 years17.
- Inspiratory respiratory dysfunction17.
- Severe autonomic failure within the first 5 years, including orthostatic hypotension or severe urinary retention/incontinence17.
- Recurrent (>1/year) falls within 3 years of onset17.
- Disproportionate anterocollis or contractures of hand or feet within the first 10 years17.
- Absence of any response to high-dose levodopa despite at least moderate severity of disease17.
- Unexplained pyramidal tract signs17.
- Bilateral symmetric parkinsonism from the onset17.
Subgroups
The subgroups of PD are not pathophysiologically different, but are used to describe which symptoms the patient presented with first:
- Tremulous (Tremor predominant)
- Akinetic Rigidity or Postural Instability and Gait Disturbances
Assessment
Hoehn and Yahr Scale can be used to assess symptom severity18.
- Higher H&Y scores have been linked with a more rapid cognitive decline in PD patients18.
- In general, patients with higher H&Y scores report poorer quality of life18.
Sleep
- Parkinson Disease Sleep Scale
Considerations
Symptom Treatment
Interventions
No current treatments can stop or reverse the neurodegenerative process5.
Community-Based Exercise
- High level of evidence and recommendation by APTA’s CPG19
- studies that improved nonmotor symptoms all included interventions for breathing and relaxation, with frequency and duration ranging from 1 to 2 hours per week for 8 to 25 weeks.
Motor Control
- Facilitate Bigger amplitude
Rehabilitation
Resources
- DPT 835 recommended readings
- O’Sullivan – CH 18
- Blumenfeld – CH 16; page 251-254
- Shumway-Cook – CH 11, 15
- Neuroanatomy: Blumenfield 2022
- Continuum:
- Parkinson Disease(Zesiewicz 2019)
- Textbook
- Localization in clinical neurology
- CPG:
- Physical Therapist Management of Parkinson Disease: A Clinical Practice Guideline From the American Physical Therapy Association (Osborne et al. 2022)
References
1.
Fulk GD, Chui KK, eds. O’Sullivan and Schmitz’s Physical Rehabilitation. 8th ed. F. A. Davis Company; 2024.
2.
Goodman C, Fuller K. Goodman and Fuller’s Pathology. 5th ed. Elsevier, Inc; 2020.
3.
Kryger MH, Roth T, Goldstein CA. Kryger’s Principles and Practice of Sleep Medicine - E-book. Elsevier Health Sciences; 2021. https://books.google.com/books?id=yDVVEAAAQBAJ
4.
Montalbán-Rodríguez A, Abalo R, López-Gómez L. From the Gut to the Brain: The Role of Enteric Glial Cells and Their Involvement in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2024;25(2):1294. doi:10.3390/ijms25021294
5.
Muleiro Alvarez M, Cano-Herrera G, Osorio Martínez MF, et al. A Comprehensive Approach to Parkinson’s Disease: Addressing Its Molecular, Clinical, and Therapeutic Aspects. International Journal of Molecular Sciences. 2024;25(13):7183. doi:10.3390/ijms25137183
6.
Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Research Reviews. 2018;42:72-85. doi:10.1016/j.arr.2017.12.007
7.
Elbaz A, Grigoletto F, Baldereschi M, et al. Familial aggregation of Parkinson’s disease: A population-based case-control study in Europe. Neurology. 1999;52(9):1876-1876. doi:10.1212/WNL.52.9.1876
8.
Morris HR, Spillantini MG, Sue CM, Williams-Gray CH. The pathogenesis of Parkinson’s disease. The Lancet. 2024;403(10423):293-304. doi:10.1016/S0140-6736(23)01478-2
9.
Skou LD, Johansen SK, Okarmus J, Meyer M. Pathogenesis of DJ-1/PARK7-Mediated Parkinson’s Disease. Cells. 2024;13(4):296. doi:10.3390/cells13040296
10.
Ulrich H. Purinergic Signaling in Neurodevelopment, Neuroinflammation and Neurodegeneration. 1st ed. Springer International Publishing AG; 2023.
11.
O’Sullivan SB, Schmitz TJ, Fulk GD, eds. Physical Rehabilitation. 7th ed. F.A. Davis Company; 2019.
12.
Breen DP, Halliday GM, Lang AE. Gut–brain axis and the spread of \(\alpha\)-synuclein pathology: Vagal highway or dead end? Movement Disorders. 2019;34(3):307-316. doi:10.1002/mds.27556
13.
Hong CT, Chen KY, Wang W, et al. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of \(\alpha\)-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells. 2020;9(3):740. doi:10.3390/cells9030740
14.
Ruiz-Pozo VA, Tamayo-Trujillo R, Cadena-Ullauri S, et al. The Molecular Mechanisms of the Relationship between Insulin Resistance and Parkinson’s Disease Pathogenesis. Nutrients. 2023;15(16):3585. doi:10.3390/nu15163585
15.
Sharma T, Kaur D, Grewal AK, Singh TG. Therapies modulating insulin resistance in Parkinson’s disease: A cross talk. Neuroscience Letters. 2021;749:135754. doi:10.1016/j.neulet.2021.135754
16.
Wang SY, Wu SL, Chen TC, Chuang CS. Antidiabetic Agents for Treatment of Parkinson’s Disease: A Meta-Analysis. International Journal of Environmental Research and Public Health. 2020;17(13):4805. doi:10.3390/ijerph17134805
17.
Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease: MDS-PD Clinical Diagnostic Criteria. Movement Disorders. 2015;30(12):1591-1601. doi:10.1002/mds.26424
18.
Modestino EJ, Reinhofer A, Blum K, Amenechi C, O’Toole P. Hoehn and Yahr staging of Parkinson’s disease in relation to neuropsychological measures. Frontiers in Bioscience (Landmark Edition). 2018;23(7):1370-1379. doi:10.2741/4649
19.
Osborne JA, Botkin R, Colon-Semenza C, et al. Physical Therapist Management of Parkinson Disease: A Clinical Practice Guideline From the American Physical Therapy Association. Physical Therapy. 2022;102(4):pzab302. doi:10.1093/ptj/pzab302
Citation
For attribution, please cite this work as:
Yomogida N, Kerstein C. Parkinson’s Disease
(PD). https://yomokerst.com/The
Archive/Neuroscience/Neuropathology/Parkinsons
Disease/parkinsons_disease.html